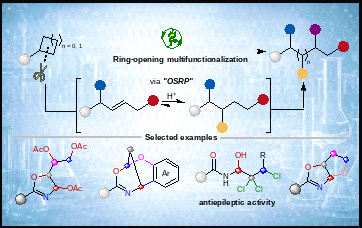
在化学世界里,环丙烷、环丁烷这类“张力环”就像被压紧的弹簧,蕴藏着可观的反应驱动力。但也有一个长期未解的难题:开环之后,反应往往只停留在1,3-或1,4-双官能团化阶段,想在相邻碳位继续“做加法”就会变得异常困难。
宁波东方理工大学副教授朱宸团队与上海交通大学副教授申涛团队等合作者,成功开发出一种可编程、发散式的电化学策略,首次实现了张力环的多位点开环多官能团化。对于未来药物和功能分子的合成而言,这意味着:化学家可以从更简单的张力环出发,更高效地“长”出过去难以一步获得的复杂结构。日前,相关成果发表在国际化学顶级期刊《自然-化学》(Nature Chemistry)上。
为什么“多改一步”这么难?
张力环开环反应是构建复杂分子的常用工具。问题在于,一旦完成初始官能团化,周围碳氢键(C–H键)的反应活性往往会明显下降,后续要在多个惰性碳-氢键 /碳-碳键(C–H/C–C键)之间同时控制“在哪儿反应、朝哪个方向反应、氧化到什么程度”,难度非常高。然而,连续碳-氧(C–O)、碳-氮(C–N)和碳-卤(C–卤键)骨架,又广泛存在于天然产物、药物分子和高值中间体中。如何让张力环从“开一次”走向“连续精准改造”,让科学家们拥有“能多改几步”的工具,一直是该领域的核心挑战。
给高活性中间体装上“节流阀”
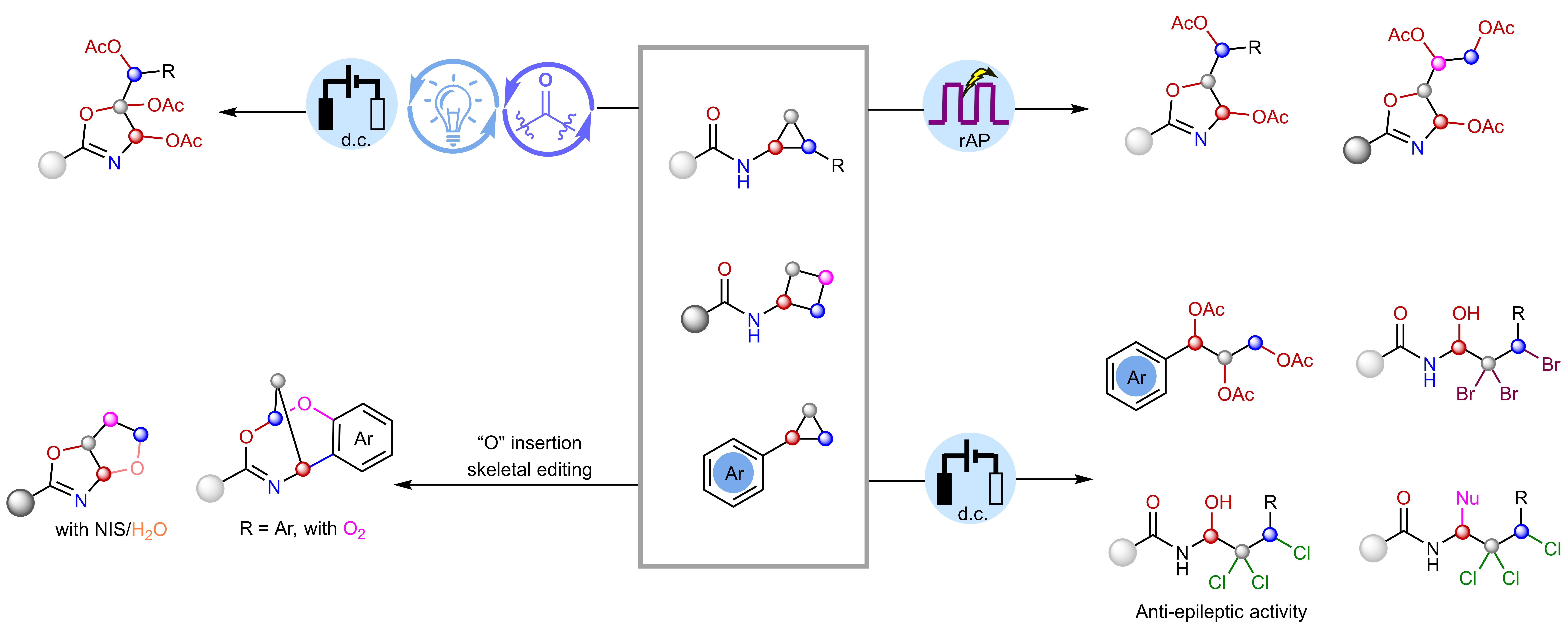
研究团队提出了一个巧妙的概念——“烯烃缓释池”(Olefin Slow-Release Pool, OSRP)。
简单来说,张力环第一次打开后,并不会一下子“失控氧化”,而是在酸性条件下缓慢、可逆地释放一种烯烃中间体,然后在电化学条件下分步完成后续氧化和官能团安装。
这就像给高活性中间体装上了节流阀,避免烯烃聚合和过度氧化,为连续活化C–C/C–H键争取了“选择性窗口”,让多步氧化反应第一次变得可控。
同一平台长出多种高价值分子
基于这一思路,研究团队将直流电解、快速交替极性电解和电光催化整合到同一个可编程平台中。通过调节酸种、阳极电位和反应模式,研究人员可以在同一类张力环底物上有选择地实现三氧化、四氧化、三卤羟化以及“O”插入骨架编辑等多种转化,把简单易得的环丙烷和环丁烷直接转化为恶唑啉、多元醇、多卤代醇及稠环骨架等高值分子。
这套方法不仅适用于不同取代模式的芳基和烷基底物,还能兼容药物相关分子片段,部分反应已实现克级放大,具备实用潜力。
DFT计算看清反应“路线图”
在这项工作中,朱宸团队承担了关键的密度泛函理论(Density functional theory, DFT)计算与机理研究。
计算表明:
反应首先在阳极氧化,生成自由基阳离子中间体,这显著拉长并削弱了张力环的碳-碳(C–C)键,推动开环。
随后发生分子内氢迁移,形成OSRP中的关键烯烃中间体。这条路径比直接消除能垒更低(23.9 vs 37.9 kcal/mol),因此反应更可控。
对于环丙酰胺体系,计算还揭示:在光敏条件下,氢原子转移优先发生在氧的邻位α-C–H(α-碳-氢键),这正是四氧化产物能够区域选择性构建的原因。
而环丁酰胺体系需要更高电位,也与计算得到的更高氧化自由能变化一致。这些结果共同勾勒出环丙烷与环丁烷氧化开环多官能团化的统一机理框架。
从“双官能化”到“多官能化”
这项研究将张力环开环化学,从传统的双官能团化,推进到了可编程、多位点、多氧化态控制的新阶段。
它不仅为高氧化态复杂分子的快速构建提供了新工具,也展示了电化学在惰性碳-碳(C–C)/碳-氢(C–H)键精准活化中的独特优势。
上海交通大学博士生李亚娟为论文第一作者,宁波东方理工大学博士后郎亚韬参与论文中的DFT计算与反应机理研究工作,朱宸与申涛为论文共同通讯作者。该研究得到了国家自然科学基金、中央高校基本科研业务费、上海交通大学2030计划、上海启源创新基金、国家科技重大专项、福建省自然科学基金等项目支持。
相关论文信息:https://doi.org/10.1038/s41557-026-02110-z
朱宸课题组介绍